

持有人和委托生产企业GMP符合性检查流程
2024-01-09 bob体育直播官网地址
属于国家管控危险化学品,根据相关法规和网安部门规定,本网站不提供该产品相关销售信息。
《药品上市后变更管理办法》对持有人的责任和义务进行了相应的规范,生产情况出现变更后的持有人应当具备符合药品生产质量管理规范的要求的生产质量管理体系,并承担药品全生命周期管理,确保药品上市后产品持续符合标准要求,并在年度报告中重点说明转让药品的情况。所以如果是持有人发生变更后要想上市销售也有必要进行符合性检查。
为什么持有人要进行符合性检查呢?持有人发生变更后,质量管理体系就会发生变更,持有人的质量管理体系变更是影响药品质量因素的主要的因素,持有人变更以后,虽然它的生产场地、生产的基本工艺没发生变更,但质量体系发生了变更,变更后的持有人能否在原药品生产场地上按照GMP的要求持续稳定的生产出与原药品一致的药品是很关键的。所以变更后的持有人必须要满足GMP符合性的要求。
《药品检查管理办法》(2021年)中相关的规定,在第三条检查程序中详细规定了检查人员的数量、检查流程以及检查方案和检查报告的要求。判定原则,该办法明确检查组一般由两名以上成员组成。检查给施行组长负责制,检查组应当按照检查方案实行现场检查,这是目前相关的法律和法规中对符合检查的相关规定。
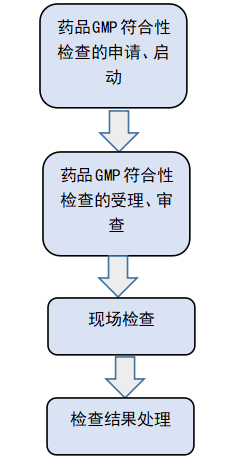
首先是审查的申请和启动,检查之前申请人一定要按照药品生产质量管理规范符合性检查申请表和符合性检查申请清单来准备相关的材料,材料准备齐全以后向省局有关部门提交,有些省局是通过它们的审批系统,有些是通过电子邮件,在提交资料之前要跟相应的省局人员进行一个沟通,确认完以后根据相关要求进行一个提交。
以笔者所在部门最近一次(2023.04)GMP检查的申请材料为例,主要有14项:
第二项生产企业许可证营业执照,这个不仅在这一项资料里面要提交持有人的和委托生产企业的。
第三项提交的是药品生产管理和质量自查情况,包括企业概况、历史沿革、上市后监督活动、关键人员和生产品种有没发生变化、上一次检查不合格项目的整改情况。
第四项资料是药品生产企业的组织结构图,要涵盖持有人的同时涵盖药品生产企业的。
第五项资料是药品生产企业法定代表人、企业负责人、生产负责人、质量负责人、质量授权人及部门负责人简历。企业负责人一定要是专职的,如果你企业负责人同时也担任别的企业的负责人的话肯定是不行的。
第六项是药品生产企业的生产范围和品种,申请检查的范围有注册证书有关的资料的复印件。
第八项是车间概况,包括所在建筑物的每层用途,车间布局、建筑面积、送回风系统、人流和物流方向、生产设备确认及验证情况、人员培训情况。
第九项是申请品种的工艺流程图,流程图上必须要明确关键的工序,主要设备包括设备的型号和规格。
第十项是生产和检验的设备,包括制水系统、空气净化系统的确认与验证情况,计算机化系统的验证情况,申请品种的工艺验证情况、清洁验证情况。
第十二项是药品生产管理和质量管理文件,这也是生产企业和持有人都必须要提供的。
第二个是受查审理,因每个省局受理的方式不一样,受理的地方也不一样,大部分是在省局的化药监管处负责接收申请,收到申请材料以后首先进行一个形式审查,如果形式审查没问题就会出具一个审查意见表。如果你的资料不完整,会叫你继续补充资料,直到符合形式审查要求以后,才将资料转交各省药品审评查验中心,中心依据相应的检查计划来制定自己的核查方案的时间。如果委托双方都在同一个省,持有人就像自己所在地的药监部门提交就行了,同时受托方应当配合提供相关的资料,省局对受托方进行一定的现场检查。如果双方不在同一个省,持有人将自己的申报资料提交自己所在地的省局,同时受托方将自己的材料提交给受托方所在的省局,最终所有的材料都要提交到持有人所在的省局。
审查受理完了以后就是现场检查,分为检查前、检查中、检查后三个阶段,检查之前核验中心必须要确定检查人员和时间,这时企业与检查组长进行一个详细的沟通,沟通的目的是确定生产计划,确定核查是为动态核查,确保所有的生产工序都能够看的到。检查组人员构成由GMP专家和检验方面的专家组成。
检查之前必须要确定检查方案,然后就是被核查单位准备好相应的申报材料,准备相应的文件清单和目录,首次会议的电脑、打印机等设备。首次会议的PPT也需要大家用心准备,其中有几项关键内容要涵盖,持有人的相应信息、受托生产企业的信息、品种信息以及持有人简介,重点要介绍持有人的质量管理体系对于上市产品全生合周期的质量保证,确定保证产品上市后的一个安全性。检查之前,企业要做好自检,确定主要的迎检人员,人员分工要明确,陪同人员回答问题必须要有一定的专业素养,能够为回答检查组提出的一些相关的问题,若遇到检查组提的问题你不知道的情况下,要找有关人员解释。
现场检查中,检查组到在单位后会张贴现场检查公示,召开首次会议,宣读检查纪律,同时双方宣读廉政承诺书,所有参会人员要签到。PPT汇报完了以后,检查人员会进行一个文件检查、生产现场和检验现场检查,这个检查会穿插着进行,检查组一般会按照检查方案实施,在检查过程中如果有些问题发生意见不一致的时候,可以和检查组做沟通并同时拿出相应的证明材料,验证自己所说的是有根据的。
最后就是检查结论,检查结果处理包括现场检查结论和综合评定结论符合标准要求、基本符合和不符合标准要求三种。药品生产监管处对《药品GMP符合性检查综合评定报告书》进行审核,综合评定为符合标准要求和基本符合标准要求的,以《药品GMP符合性检查结果通知书》方式通知被检查单位。综合评定结论为不符合标准要求的,《药品GMP符合性检查意见书》方式告知被检查单位。
药品生产监管处会负责对药品GMP符合性检查结果公示,有省局网站发布《药品GMP符合性检查结果公告》。在这里提示警醒我们要重视你的GMP检查结论,因为你单位的每一次检查都会纳入企业信用档案,将来对对持有人生产和其它检查都会产生比较大的影响。
更多
本周,有不少热点需要我们来关注。审评审批方面,多个药物获批。最值...[详细]
2023年12月22日,中国食品药品检定研究院(以下简称中检院)官网...[详细]
全球首 款PD-1/CTLA-4双抗申报新适应症,一线日,康方生物「卡度尼利单抗注射液」的2.2类注册申请获CDE受...[详细]
过去的一年,据NMPA官网批件信息统计,共有80余款新药在国内首次...[详细]
12月11日,FDA公布了一则召回通告。InvaGen Pharmaceuticals的一款氨己烯...[详细]